
(Epi)Genetic Profiling Services
Overview
Please Note:
We have changed our sample drop off protocols and times - click on and read the following PDFs for details:
- The core accepts samples that require library preparation and sequencing. There will be a quality control step prior to agreement of service. Please refer to specific services using the menu on the left for detailed quality control descriptions.
- The price for library preparation is per library, and includes sample quality control, library preparation and library validation.
- The price for sequencing varies based on sample volume and number of reads required per sample, and includes de-multiplexing (if required), post-processing (if available and requested) and two years data storage. For methylation sequencing services, methylation calls are also provided with alignments as part of the post-processing.
- Sequencing is performed on NovaSeq 6000 or MiSeq Illumina instruments.
- A Bioinformatics Fee (10% of the sequencing price for Internal Clients and 20% for External Clients) will be added.
- Libraries made by the core, routinely yield clusters between 750-800 k/mm2. We cannot guarantee similar clustering and/or quality for libraries made by customers of the core.
- We require that core clients acknowledge the Epigenomics Core of Weill Cornell Medical College in publications and presentions enabled by Epigenomics core resources.
Sample submission tips
- To request a sequencing service, please fill out a request in our Agilent Crosslab/iLab Service Request LIMS. Please refer to the GETTING STARTED section of our Sample Workflow above for detailed sample submission instructions.
- Please use the Agilent Crosslab/iLab service ID and the sample number you have indicated in the submission form to label your tubes clearly, tubes without a service ID and sample number will not be accepted.
- Once samples are submitted and pass quality control, they are entered into a sample or library queue.
- The MiSeq personal sequencer has a single lane and only supports paired end clustering. [MiSeq PDF]
Timeline:
Approximate time for library preps and sequencing is 4-8 weeks if all samples pass quality control.
Approximate time for data pre-processing and transfer is 1-2 weeks from the end of a successful sequencing run.
RRBS (DNA Methylation Sequencing)
Assay Description
Reduced Representation Bisulfite Sequencing is a modification of the original RRBS protocol (Gu H. et al. 2011) and the in-house developed ERRBS method (Akalin A., Garrett-Bakelman F. et al, 2012) for base-pair resolution methylation sequencing analysis based on the use of a restriction enzyme to enrich for CpG fragments. RRBS starts with MspI digestion, followed by NGS-library preparation and bisulfite conversion of cytosines.
Our RRBS protocol yields about ~10% of genomic CpG sites (roughly 3M CpGs in the human genome), and provides enrichment in CpG islands and CpG shores, promoters, exons, introns and intergenic regions (Garrett-Bakelman F., Sheridan, C. et al. 2015).
This assay requires RNA-free high molecular weight DNA. Degraded DNA, such as that obtained from FFPE is not suitable. FFPE samples may be processed using Agilent's Methylome Capture assay instead.
Sample Requirement:
Submit 75 ng of genomic DNA, of molecular weight >20kb, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2; RNA-free and at a concentration of ~20ng/ul
Epigenomics Core Quality Control:
Determination of concentration of double stranded DNA (dsDNA) using Qubit Fluorometer, Agilent Tape station 4200 or agarose gel to determine molecular weight.
[Click here for a detailed description of the QC]
100 million (M) read per sample on a paired end read flow cell with 100bp read length (PE100) are recommended for differential methylation analysis, for detailed information please review Garrett-Bakelman F., Sheridan, C. et al. 2015.
Third Party Resource: RRBS Guide from Babraham Bioinformatics (makers of the Bismark aligner, FASTQC, Seqmonk etc.)
Some studies that have used our RRBS assay: Odell SC. et al. 2020, Emi T. et al. 2020
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Library Prep | $205 |
| NovaSeq X 200 cycles 1.25B Reads + Bioinformatics processing | $1262 |
Prices may vary depending on the number of samples.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
We process Bisulfite sequencing data using an in-house BWA meth pipeline to generate methylation calls, related statistics and bam files for the samples.
|
Sample_CpG.methylKit.gz Sample_CHH.methylKit.gz Sample_CHG.methylKit.gz |
The Sample_*.methylKit.gz files are tab delimited text files that contain all the reported locations of C's in either CpG, CHG, or CHH, context and their methylation levels. The minimum read coverage cutoff for this file is 1 |
|---|---|
|
cpg.Sample.10x.gz chh.Sample.10x.gz chg.Sample.10x.gz |
The *.10x.txt.gz files are tab delimited text files that contain the locations of C's in either CpG, CHG, or CHH, context and their methylation levels with a coverage greater than or equal to 10 (>=10x) |
| Sample_sorted.bam Sample_sorted.bam.bai |
This file contains the complete alignments in binary (BAM) format and a index (.bai) for this BAM file. |
| Sample_summary.txt | This file summarizes adapter trimming, alignment information, and mapping efficiency of the sample against the genome. |
The column headers for the *.methylkit.gz and *.10x.gz are as follows:
- chrBase = This is the name (chromosome.base location)
- chr = chromosome on which the methylated base is located
- base = location of methylated base on the chromosome
- strand = forward strand (F) or reverse strand (R)
- coverage = read coverage
- freqC = % methylated
- freqT = % unmethylated
Please refer to the following paper for more information about BWA meth: Fast and accurate alignment of long bisulfite-seq reads. Pedersen, BS et al. (2014) ArXiv.
RR(Ox)BS (5hmC Sequencing)
Assay Description
Reduced Representation Oxidative Bisulfite Sequencing enables an accurate 5-hydroxymethylcytosine (5hmC) identification. Please note that the protocol requires two library preparations per sample. The core uses methodology described in Akalin A., Garrett-Bakelman F. et al, 2012 and Garrett-Bakelman F., Sheridan, C. et al. 2015), with the chemistry from Tecan’s Ultralow Methyl-Seq with TrueMethyl oxBS to prepare the libraries.
Sample Requirement:
Submit 400 ng of genomic DNA, of molecular weight >20kb, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2; RNA-free and at a concentration of ~20ng/ul. Due to sequencing recipe requirements please submit a minimum of 4 samples (8 libraries).
Epigenomics Core Quality Control:
Determination of concentration of double stranded DNA (dsDNA) using Qubit Fluorometer, Agilent Tape station 4200 or agarose gel to determine molecular weight.
[Click here for a detailed description of the QC]
100 million (M) reads per library on a single end read flow cell with 100 sequencing cycles (SR100) are recommended for differential methylation analysis, for detailed information please review Garrett-Bakelman F., Sheridan, C. et al. 2015.
Some studies that have used our RRoxBS assay: Singh P. et al. 2021, Fortin J. et al. 2023
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Library Prep (per sample) | $255 |
| NextSeq 2000 (400M reads) Single Read 100bp (SR100) + Bioinformatics Processing | $1980 |
| NovaSeq X 100 cycles 1.25B Reads + Bioinformatics Processing | $1742 |
Prices may vary depending on the number of samples.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
We process Bisulfite sequencing data using an in-house BWA meth pipeline to generate methylation calls, related statistics and bam files for the samples.
|
Sample_CpG.methylKit.gz Sample_CHH.methylKit.gz Sample_CHG.methylKit.gz |
The Sample_*.methylKit.gz files are tab delimited text files that contain all the reported locations of C's in either CpG, CHG, or CHH, context and their methylation levels. The minimum read coverage cutoff for this file is 1 |
|---|---|
|
cpg.Sample.10x.gz chh.Sample.10x.gz chg.Sample.10x.gz |
The *.10x.txt.gz files are tab delimited text files that contain the locations of C's in either CpG, CHG, or CHH, context and their methylation levels with a coverage greater than or equal to 10 (>=10x) |
| Sample_sorted.bam Sample_sorted.bam.bai |
This file contains the complete alignments in binary (BAM) format and a index (.bai) for this BAM file. |
| Sample_summary.txt | This file summarizes adapter trimming, alignment information, and mapping efficiency of the sample against the genome. |
The column headers for the *.methylkit.gz and *.10x.gz are as follows:
- chrBase = This is the name (chromosome.base location)
- chr = chromosome on which the methylated base is located
- base = location of methylated base on the chromosome
- strand = forward strand (F) or reverse strand (R)
- coverage = read coverage
- freqC = % methylated
- freqT = % unmethylated
Please refer to the following paper for more information about BWA meth: Fast and accurate alignment of long bisulfite-seq reads. Pedersen, BS et al. (2014) ArXiv.
Whole Genome Bisulfite Sequencing (WGBS) - Genome-wide methylation sequencing
Assay Description
Whole genome bisulfite sequencing is the gold-standard approach for comprehensive base-pair resolution and quantitative information at most genomic cytosines. The core uses the Accel-NGS Methyl-Seq DNA library kit to prepare libraries. This method uses cytosine bisulfite conversion followed by single stranded ligation of adapters and PCR amplification. [Application Note].
Sample Requirement:
Submit 300 ng of genomic DNA, of molecular weight >20kb, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2; RNA-free and at a concentration of ~20ng/ul
Epigenomics Core Quality Control:
Determination of concentration of double stranded DNA (dsDNA) using Qubit Fluorometer, Agilent Tape station 4200 or agarose gel to determine molecular weight.
[Click here for a detailed description of the QC]
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Library Prep | $260 |
To calculate the sequencing depth required for your desired coverage and species of interest, please use the Illumina coverage calculator.
Estimate pricing as NovaSeq X 300 cycles 1.25B Reads + Bioinformatics processing = $1,345
Sequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
We process Bisulfite sequencing data using an in-house BWA meth pipeline to generate methylation calls, related statistics and bam files for the samples.
|
Sample_CpG.methylKit.gz Sample_CHH.methylKit.gz Sample_CHG.methylKit.gz |
The Sample_*.methylKit.gz files are tab delimited text files that contain all the reported locations of C's in either CpG, CHG, or CHH, context and their methylation levels. The minimum read coverage cutoff for this file is 1 |
|---|---|
|
cpg.Sample.10x.gz chh.Sample.10x.gz chg.Sample.10x.gz |
The *.10x.txt.gz files are tab delimited text files that contain the locations of C's in either CpG, CHG, or CHH, context and their methylation levels with a coverage greater than or equal to 10 (>=10x) |
| Sample_sorted.bam Sample_sorted.bam.bai |
This file contains the complete alignments in binary (BAM) format and a index (.bai) for this BAM file. |
| Sample_summary.txt | This file summarizes adapter trimming, alignment information, and mapping efficiency of the sample against the genome. |
The column headers for the *.methylkit.gz and *.10x.gz are as follows:
- chrBase = This is the name (chromosome.base location)
- chr = chromosome on which the methylated base is located
- base = location of methylated base on the chromosome
- strand = forward strand (F) or reverse strand (R)
- coverage = read coverage
- freqC = % methylated
- freqT = % unmethylated
Please refer to the following paper for more information about BWA meth: Fast and accurate alignment of long bisulfite-seq reads. Pedersen, BS et al. (2014) ArXiv.
Whole Genome Oxidative Bisulfite Sequencing (WGoxBS) - Genome-wide 5hmC sequencing
Assay Description
Whole Genome Oxidative Bisulfite Sequencing (WGoxBS) enables an accurate 5-methylcytosine (5mC) identification and interrogation of both 5-hydroxymethylcytosine (5hmC) and 5-methylcytosine (5mC). Please note that the protocol requires two library preparations per sample, and therefore twice the sequencing depth than WGBS. The core uses Tecan’s Ultralow Methyl-Seq with TrueMethyl oxBS kit to prepare libraries.
Sample Requirement:
Submit 400 ng of genomic DNA, of molecular weight >20kb, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2; RNA-free and at a concentration of ~20ng/ul. Due to sequencing recipe requirements please submit a minimum of 6 samples (12 libraries).
Epigenomics Core Quality Control:
Determination of concentration of double stranded DNA (dsDNA) using Qubit Fluorometer, Agilent Tape station 4200 or agarose gel to determine molecular weight.
[Click here for a detailed description of the QC]
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Library Prep | 350 |
To calculate the sequencing depth required for your desired coverage and species of interest, please use the Illumina coverage calculator.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
We process Bisulfite sequencing data using an in-house BWA meth pipeline to generate methylation calls, related statistics and bam files for the samples.
|
Sample_CpG.methylKit.gz Sample_CHH.methylKit.gz Sample_CHG.methylKit.gz |
The Sample_*.methylKit.gz files are tab delimited text files that contain all the reported locations of C's in either CpG, CHG, or CHH, context and their methylation levels. The minimum read coverage cutoff for this file is 1 |
|---|---|
|
cpg.Sample.10x.gz chh.Sample.10x.gz chg.Sample.10x.gz |
The *.10x.txt.gz files are tab delimited text files that contain the locations of C's in either CpG, CHG, or CHH, context and their methylation levels with a coverage greater than or equal to 10 (>=10x) |
| Sample_sorted.bam Sample_sorted.bam.bai |
This file contains the complete alignments in binary (BAM) format and a index (.bai) for this BAM file. |
| Sample_summary.txt | This file summarizes adapter trimming, alignment information, and mapping efficiency of the sample against the genome. |
The column headers for the *.methylkit.gz and *.10x.gz are as follows:
- chrBase = This is the name (chromosome.base location)
- chr = chromosome on which the methylated base is located
- base = location of methylated base on the chromosome
- strand = forward strand (F) or reverse strand (R)
- coverage = read coverage
- freqC = % methylated
- freqT = % unmethylated
Please refer to the following paper for more information about BWA meth: Fast and accurate alignment of long bisulfite-seq reads. Pedersen, BS et al. (2014) ArXiv.
Methylome Capture Sequencing (targeted methylome sequencing)
Assay Description
Methylcapture is a hybridization-based approach on platforms containing pre-designed capture oligos, followed by methylation sequencing. There are commercially designed capture libraries available with a range
of epigenetic features that cover ~12% to ~24% of all human genome CpGs [Agilent SureSelect MethylSeq | Roche CpGiant].
Please contact us if you would like to use this service.
Advantages:
- Researchers can custom-design capture libraries that can be used for validation or for discovery of novel epigenomic regions. Please contact us if you would like to design a custom library.
- Amenable for FFPE samples. Since the technique relies on a sonication step, FFPE DNA can be used.
Sample Requirement:
Depending on the platform used, submit 0.5-2ug of genomic DNA, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2, RNA-free and at a concentration of ~50ng/ul
Epigenomics Core Quality Control:
Determination of concentration of double stranded DNA (dsDNA) using Qubit Fluorometer, Perkin Elmer Labchip GX or agarose gel to determine molecular weight.
[Click here for a detailed description of the QC]
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Library Prep | 425 |
PE100 (2x100 cycles) sequencing is recommended. Depth depends on the chosen commercially available capture panel. To calculate the sequencing depth required for you panel/species, please use the Illumina coverage calculator.
The same data processing pipeline we use for our RRBS assay will be used for methylome capture data as well.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
We process Bisulfite sequencing data using an in-house BWA meth pipeline to generate methylation calls, related statistics and bam files for the samples.
|
Sample_CpG.methylKit.gz Sample_CHH.methylKit.gz Sample_CHG.methylKit.gz |
The Sample_*.methylKit.gz files are tab delimited text files that contain all the reported locations of C's in either CpG, CHG, or CHH, context and their methylation levels. The minimum read coverage cutoff for this file is 1 |
|---|---|
|
cpg.Sample.10x.gz chh.Sample.10x.gz chg.Sample.10x.gz |
The *.10x.txt.gz files are tab delimited text files that contain the locations of C's in either CpG, CHG, or CHH, context and their methylation levels with a coverage greater than or equal to 10 (>=10x) |
| Sample_sorted.bam Sample_sorted.bam.bai |
This file contains the complete alignments in binary (BAM) format and a index (.bai) for this BAM file. |
| Sample_summary.txt | This file summarizes adapter trimming, alignment information, and mapping efficiency of the sample against the genome. |
The column headers for the *.methylkit.gz and *.10x.gz are as follows:
- chrBase = This is the name (chromosome.base location)
- chr = chromosome on which the methylated base is located
- base = location of methylated base on the chromosome
- strand = forward strand (F) or reverse strand (R)
- coverage = read coverage
- freqC = % methylated
- freqT = % unmethylated
Please refer to the following paper for more information about BWA meth: Fast and accurate alignment of long bisulfite-seq reads. Pedersen, BS et al. (2014) ArXiv.
Enzymatic Methyl-Seq (EMSeq) - Genome-wide methylation sequencing
Assay Description
The Enzymatic Methyl Sequencing assay provides a high-performance enzyme-based alternative to bisulfite conversion for identification of methylation in low input samples. The core uses the NEBNext Enzymatic Methyl-seq kit to prepare libraries.
Sample Requirement:
Submit 10-50 ng of genomic DNA, of molecular weight >20kb, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2; RNA-free and at a concentration of ~20ng/ul.
Epigenomics Core Quality Control:
Determination of concentration of double stranded DNA (dsDNA) using Qubit Fluorometer, Agilent Tape station 4200 or agarose gel to determine molecular weight.
[Click here for a detailed description of the QC]
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Library Prep | 280 |
To calculate the sequencing depth required for your desired coverage and species of interest, please use the Illumina coverage calculator.
Estimate pricing as NovaSeq X 300 cycles 1.25B Reads + Bioinformatics processing = $1,345
Sequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
We process Bisulfite sequencing data using an in-house BWA meth pipeline to generate methylation calls, related statistics and bam files for the samples.
|
Sample_CpG.methylKit.gz Sample_CHH.methylKit.gz Sample_CHG.methylKit.gz |
The Sample_*.methylKit.gz files are tab delimited text files that contain all the reported locations of C's in either CpG, CHG, or CHH, context and their methylation levels. The minimum read coverage cutoff for this file is 1 |
|---|---|
|
cpg.Sample.10x.gz chh.Sample.10x.gz chg.Sample.10x.gz |
The *.10x.txt.gz files are tab delimited text files that contain the locations of C's in either CpG, CHG, or CHH, context and their methylation levels with a coverage greater than or equal to 10 (>=10x) |
| Sample_sorted.bam Sample_sorted.bam.bai |
This file contains the complete alignments in binary (BAM) format and a index (.bai) for this BAM file. |
| Sample_summary.txt | This file summarizes adapter trimming, alignment information, and mapping efficiency of the sample against the genome. |
The column headers for the *.methylkit.gz and *.10x.gz are as follows:
- chrBase = This is the name (chromosome.base location)
- chr = chromosome on which the methylated base is located
- base = location of methylated base on the chromosome
- strand = forward strand (F) or reverse strand (R)
- coverage = read coverage
- freqC = % methylated
- freqT = % unmethylated
Please refer to the following paper for more information about BWA meth: Fast and accurate alignment of long bisulfite-seq reads. Pedersen, BS et al. (2014) ArXiv.
5hmC-BIC-Seq (detection of 5-hydroxymethylcytosine modification by affinity pull down)
Assay Description
5hmC-bead-integrated-click-sequencing is an in-house method developed by the core to profile 5hmC containing DNA sequences on a genome-wide scale that uses a novel integrated approach. Typical enrichment protocols use antibodies that recognize a modification or set of modifications on DNA. We chose a covalent chemical labeling technique that could be integrated into the NGS library preparation process. DNA sequences with 5hmC moieties are directly modified with azide-glucose, which can then form a stable biotin conjugate through bio orthogonal click-chemistry. Streptavidin affinity purification enriches 5hmC-containing DNA sequences and integration with sample preparation steps creates a robust assay that can accept limiting levels of input DNA.
Please contact us if you wish to use this service.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
We process Bisulfite sequencing data using an in-house BWA meth pipeline to generate methylation calls, related statistics and bam files for the samples.
|
Sample_CpG.methylKit.gz Sample_CHH.methylKit.gz Sample_CHG.methylKit.gz |
The Sample_*.methylKit.gz files are tab delimited text files that contain all the reported locations of C's in either CpG, CHG, or CHH, context and their methylation levels. The minimum read coverage cutoff for this file is 1 |
|---|---|
|
cpg.Sample.10x.gz chh.Sample.10x.gz chg.Sample.10x.gz |
The *.10x.txt.gz files are tab delimited text files that contain the locations of C's in either CpG, CHG, or CHH, context and their methylation levels with a coverage greater than or equal to 10 (>=10x) |
| Sample_sorted.bam Sample_sorted.bam.bai |
This file contains the complete alignments in binary (BAM) format and a index (.bai) for this BAM file. |
| Sample_summary.txt | This file summarizes adapter trimming, alignment information, and mapping efficiency of the sample against the genome. |
The column headers for the *.methylkit.gz and *.10x.gz are as follows:
- chrBase = This is the name (chromosome.base location)
- chr = chromosome on which the methylated base is located
- base = location of methylated base on the chromosome
- strand = forward strand (F) or reverse strand (R)
- coverage = read coverage
- freqC = % methylated
- freqT = % unmethylated
Please refer to the following paper for more information about BWA meth: Fast and accurate alignment of long bisulfite-seq reads. Pedersen, BS et al. (2014) ArXiv.
ChIPseq (genome-wide mapping of DNA binding proteins)
Assay description
Chromatin Immunoprecipitation Sequencing (Carey et al, 2009 http://cshprotocols.cshlp.org/) is the primary method for profiling protein DNA interactions, based on the enrichment of DNA associated to a protein of interest or a histone modification. Despite its increasing use ChIP-seq libraries are among the most challenging to perform and require significant experience and quality control measures. The core offers library preparation for chromatin immunoprecipitated material after successful quality control.
Sample Requirement:
For Input material submit 50 ng at a concentration of 5ng/ul.
For ChIP material submit ~22ng of DNA at a concentration of 0.3ng/ul to 1ng/ul.
Please use a double stranded fluorometric method to determine concentration (Qubit for example); concentrations determined by nanodrop are not reliable for this assay.
Epigenomics Core Quality Control:
QC1 - Quantity: dsDNA using Qubit Fluorometer.
QC2 - Quality: High Sensitivity DNA Bioanalyzer chip to determine the spread of the Input chromatin. Accurate representation of the original ChIPd material is obtained when the size range of the DNA fraction required for library preparation (130-230bp) is > 10% of the total DNA provided.
[Click here for a detailed description of the QC]
Library preparation for this assay is currently done using the IDT-xGen kit.
Some studies that have used our ChIPSeq sequencing service: Popovic R. et al. 2014, Kuo PY. et al. 2014, Qiao Y. et al. 2013
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| IDT-xGen ChIP-seq Library Prep | $180 |
| NextSeq 2000 (400M reads) Paired End 50bp (PE50) + Bioinformatics Processing | 1980 |
| NovaSeq X 100 cycles 1.25B Reads + Bioinformatics processing | $1742 |
50M reads of paired end 50 (PE50) sequencing is recommended per sample. Multiplexing optimization depends on the factor used, for well characterized antibodies or sharp histone marks (for example: H3K4me3), 25 million reads may be sufficient per sample.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
FASTQ files generated as described above are aligned to genomes available via Illumina's iGenome using the BWA-MEM aligner. This pipeline results in the following file types:
*.maxL.bam - The top/best non-filtered alignment for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.merged.bam - all alignments (including best and multiple alignments) for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.maxL.bam.bai, *.merged.bam.bai - Index files (.bai files) for each sample which allow for easier viewing of the bam files in genome browsers such as UCSC Genome Browser) or IGV).
*-metrics.log - Summary metrics such as adapter trimming and alignment rates.
ATAC-seq (assay for transposase activity)
Assay description
Assessment of the functional state of chromatin, can be achieved through the digestion of chromatin with Tn5 transposase followed by library preparation and sequencing. The Epigenomics core uses the OMNI-ATAC protocol as detailed in Corces et al. (Nature Methods, 2017)
Sample Requirement:
After submitting a service request in Agilent Crosslab (formerly iLab),
please follow the protocol detailed in this PDF, label the Eppendorf tube with the Agilent CrossLab ID and the sample number (as in the service request) and bring samples after tagmentation to room A-427. We do not accept cells for ATAC-seq.
Instructions for preparation of OMNI ATAC-seq samples [PDF]
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| ATAC-seq (Omni) Library Prep (Tagmented Samples) | $105 |
| NovaSeq X 100 cycles 1.25B Reads + Bioinformatics processing | $996 |
50M reads of paired end 50 (PE50) sequencing is recommended per sample.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
FASTQ files generated as described above are aligned to genomes available via Illumina's iGenome using the BWA-MEM aligner. This pipeline results in the following file types:
*.maxL.bam - The top/best non-filtered alignment for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.merged.bam - all alignments (including best and multiple alignments) for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.maxL.bam.bai, *.merged.bam.bai - Index files (.bai files) for each sample which allow for easier viewing of the bam files in genome browsers such as UCSC Genome Browser) or IGV).
*-metrics.log - Summary metrics such as adapter trimming and alignment rates.
Exome Capture
Assay description
The Agilent SureSelect platform is a solution-based system using 120-mer (biotinylated cRNA baits) to capture regions of interest, enriching them out of a NGS sonicated genomic library. Targeted regions can be interrogated for the purpose of identifying structural rearrangements and SNPs. [SureSelect PDF]
Sample Requirement:
Please submit a minimum of 12 samples. Submit 3ug of purified DNA, Nanodrop A260/280 ratio> 1.7; A260/230 =2.0-2.2
Epigenomics Core Quality Control:
dsDNA using Qubit Fluorometer, 0.8 % agarose gel to determine quality
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
External Academic Price |
|---|---|---|
| Exome Capture TruSeq Compatible Library Prep | 528 | 600 |
| HiSeq PE75 Paired End Clustering and 3 x 50 Sequencing Cycles | 2050 | 3000 |
1 lane of PE75 sequencing is recommended for commercially available capture libraries. To calculate the sequencing depth required for you species of interest, please use the Illumina coverage calculator.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
FASTQ files generated as described above are aligned to genomes available via Illumina's iGenome using the BWA-MEM aligner. This pipeline results in the following file types:
*.maxL.bam - The top/best non-filtered alignment for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.merged.bam - all alignments (including best and multiple alignments) for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.maxL.bam.bai, *.merged.bam.bai - Index files (.bai files) for each sample which allow for easier viewing of the bam files in genome browsers such as UCSC Genome Browser) or IGV).
*-metrics.log - Summary metrics such as adapter trimming and alignment rates.
DNASeq (whole genome sequencing)
To continue availing of this service, please contact Dr. Jenny Xiang at the Genomics Resources Core Facility (GRCF).
[GRCF Service Request Portal]
An entire genome can be sequenced allowing SNP discovery, identification of copy number variations and chromosomal rearrangements [Nanokit]
Sample Requirement:
Submit 250 ng of genomic DNA, of molecular weight >40kb, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2; RNA-free and at a concentration of ~20ng/ul
Epigenomics Core Quality Control:
dsDNA using Qubit Fluorometer, 0.8 % agarose gel to determine quality
[Click here for a detailed description of the QC]
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| TruSeq DNA-seq Library Prep | $300 |
Two lanes of PE75 or PE100 sequencing recommended for human DNAseq. To calculate depth required for your custom application, please use the Illumina coverage calculator.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
FASTQ files generated as described above are aligned to genomes available via Illumina's iGenome using the BWA-MEM aligner. This pipeline results in the following file types:
*.maxL.bam - The top/best non-filtered alignment for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.merged.bam - all alignments (including best and multiple alignments) for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.maxL.bam.bai, *.merged.bam.bai - Index files (.bai files) for each sample which allow for easier viewing of the bam files in genome browsers such as UCSC Genome Browser) or IGV).
*-metrics.log - Summary metrics such as adapter trimming and alignment rates.
Targeted Resequencing (interrogation of genes specific for heme malignancies)
Assay description
Using Raindance’s single-molecule pico droplet PCR reaction three panels that contain genes specific for heme malignancies have been designed:
- Myeloid leukemia
- Chronic Lymphocytic Leukemia
- Lymphoma
This is a high-throughput technique, a minimum of 40 samples are required.
Sample Requirement:
Submit 500 ng of genomic DNA, of molecular weight >40kb, Nanodrop A260/280 ratio >1.7; A260/230=2.0-2.2; at ~20ng/ul
Epigenomics Core Quality Control:
Determination of concentration of double stranded DNA (dsDNA) using Qubit Fluorometer, Perkin Elmer Labchip GX or agarose gel to determine molecular weight.
| Service | Internal (WCM, WCM Qatar & Cornell U) Price |
External Academic Price |
|---|---|---|
| Raindance Library Prep | 200 | 400 |
| MiSeq 600 Sequencing Cycles v3 | 1500 | 2400 |
Please contact us if you wish to use this service.
Data ProcessingSequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
FASTQ files generated as described above are aligned to genomes available via Illumina's iGenome using the BWA-MEM aligner. This pipeline results in the following file types:
*.maxL.bam - The top/best non-filtered alignment for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.merged.bam - all alignments (including best and multiple alignments) for each read in the widely accepted BAM format (a binary version of the SAM format) for each sample.
*.maxL.bam.bai, *.merged.bam.bai - Index files (.bai files) for each sample which allow for easier viewing of the bam files in genome browsers such as UCSC Genome Browser) or IGV).
*-metrics.log - Summary metrics such as adapter trimming and alignment rates.
Single Cell Gene Expression
Assay description
Our single-cell gene expression services includes the following 10x Genomics GEM-X technology single cell gene expression assays [PDF]: single-plex, On-Chip-Multiplexing (OCM) and Flex.
Single cell assays require prior consultation and coordination with the core. Please contact us at epicore-sc-coord@med.cornell.edu at least three weeks before dropping off your samples to schedule a time. We currently only accept external customers coming from within a mile radius of our facility.
The 10x Genomics Chromium Single Cell Expression Solution provides high-throughput, single cell expression measurements that enable discovery of gene expression dynamics and molecular profiling of individual cell types. This is also available in conjunction with Feature Barcoding (Cell Surface Protein, CRISPR Screening or Custom). The protocol requires a suspension of viable single cells as input. Minimizing the presence of cellular aggregates, dead cells, non-cellular nucleic acids and potential inhibitors of reverse transcription is critical to obtaining high quality data.
Sample Requirement:
- The total number of cells required in the suspension used as input is determined by the desired cell recovery target (between 500-20000 cells for single-plex and 500-5000 individual cells for OCM); required sequencing read depth depends on the desired cell target. Please check user guides CG000731 and CG000768 for details. [Library Prep User Guides]
- For Flex we require 300,000 to 1,000,000 fixed cells. Please check user guide CG000478
- Given the variety of cells and sample types, general guidelines for sample preparation need to be optimized by each customer. Please check the 10X Genomics Single Cell Gene Expression Sample Prep Guide for information on how to prepare cells.
| Singleplex Service | Internal Price (WCM, WCM Qatar & Cornell U) |
Multiplex Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|---|---|
| Single Cell 3P Library Prep (1 sample) | $2400 | On Chip Multiplexing (OCM), (4 samples), each | $895 |
| Single Cell 3P Library Prep (2-4 samples), each | $2150 | Flex v2 Gene Expression (12 or 16 samples), each | $600 |
| Single Cell 3P Library Prep (>5 samples), each | $2050 | Flex v2 Gene Expression (4 or 8 samples), each | $1219 |
| Single Cell 3P Gene expression with Cell Hashing Library Prep (per sample) | $2550 | ||
| Additional Feature Barcoding Library Prep (per sample) | $250 | ||
| GEM-X Chip | $325 | GEM-X Chip | $325 |
| NovaSeq X (100 cycles, 1.25B reads) + Bioinformatics Processing | $996 |
We sequence Chromium single cell samples on a pair end flow cell (28-10-10-90) with a 2x50 cycles kit. Chromium recommends a minimum of 20,000 reads per cell for gene expression (Example: Samples containing from 2000-10,000 cells would require from 40-200 million reads per sample). Please contact us at epigenomicscore@med.cornell.edu to discuss the appropriate number of reads required for your experiments.
Data Processing
The sequencing data will be demultiplexed and post-processed using custom pipelines provided by 10x Genomics.
Feature Barcoding (Cell Surface Protein, CRISPR Screening or Custom) and Cell Multiplexing assays require additional information for post-processing with cellranger (for collating the feature counts with the gene expression counts).
Below please find a description of the columns in the Feature Reference table:
| Sample Name | The name you have used for the samples while submitting them in iLab |
| Feature Name | Human-readable name for this feature. Must not contain whitespace. This name will be displayed in Loupe Cell Browser. |
| Read (R1 or R2) | Specifies which RNA sequencing read contains the Feature Barocde sequence. Must be R1 or R2. Note: in most cases R2 is the correct read. |
| Pattern / Position of Barcode | The pattern column can be made up of a combination of these elements: 5P: denotes the beginning of the read sequence. May appear 0 or 1 times, and must be at the beginning of the pattern. Only 5P or 3P may appear, not both. 3P: denotes the end of the read sequence. May appear 0 or 1 times, and must be at the end of the pattern. N: denotes an arbitrary base. A, C, G, T: denotes a fixed base that must match the read sequence exactly. (BC): denotes the Feature Barcode sequence as specified in the sequence column of the feature reference. Must appear exactly once in the pattern. |
| Sequence | Nucleotide barcode sequence associated with this feature. E.g., antibody barcode or sgRNA protospacer sequence. |
The 10X Genomics Feature Barcoding page has detailed information about the post processing of feature barcoding data and required input for cellranger.
Single Cell Immune Profiling
Assay description
Our single-cell gene expression services includes the following 10x Genomics GEM-X technology single cell gene expression assays [PDF]: Single plex and On-Chip-Multiplexing (OCM)
Single cell assays require prior consultation and coordination with the core. Please contact us at epicore-sc-coord@med.cornell.edu at least three weeks before dropping off your samples to schedule a time. We currently only accept external customers coming from within a mile radius of our facility.
The Chromium Single Cell Immune Profiling Solution
The Chromium Single Cell Immune Profiling Solution is a comprehensive approach to simultaneously examine the cellular context of the adaptive immune response and immune repertoires of hundreds to tens of thousands of T and B cells in human or mouse on a cell-by-cell basis. With the addition of Feature Barcoding Technology, you can now also detect and analyze additional cellular readouts such as cell surface markers and antigen specificity to enhance immune cell phenotyping and study dynamic interactions between lymphocytes and target cells.
Sample Requirement:
The protocol requires a suspension of viable single cells as input. Minimizing the presence of cellular aggregates, dead cells, non-cellular nucleic acids and potential inhibitors of reverse transcription is critical to obtaining high quality data.
- The total number of cells required in the suspension used as input is determined by the desired cell recovery target (between 500-20000 cells for single-plex and 500-5000 individual cells for OCM); required sequencing read depth depends on the desired cell target. Please check user guides CG000733 and CG000770 for details. [Library Prep User Guide]
- Given the variety of cells and sample types, general guidelines for sample preparation need to be optimized by each customer. Please check the 10X Genomics Single Cell Immune Profiling Sample Prep Guide for information on how to prepare cells.
- We can process up to 4 samples at a time.
The library preparation prices below are for the 5 prime gene expression assay as well as either a T or a B cell V(D)J assay.
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Single Cell 5P gene expression + VDJ repertoire (1 sample) | $2500 |
| On Chip Multiplexing (OCM), 4 samples, each | $895 |
| Single Cell 5P gene expression + VDJ repertoire (2-4 samples) | $2200 |
| Single Cell 5P gene expression + VDJ repertoire (>5 samples) | $2100 |
| Single Cell 5P gene expression + VDJ repertoire + Hashing | $2800 |
| Additional Immuno Profiling Feature Barcoding Library Prep (per sample) | $250 |
| GEM-X Chip | $325 |
| NovaSeq (100 cycles 1.25B Reads) + Bioinformatics Processing | $996 |
We sequence Chromium single cell samples on a pair end flow cell (28-10-10-91) with a 2x50 cycles kit. Chromium recommends 50,000 reads per cell with a minimum of 20,000 reads per cell for gene expression (Example: Samples containing from 2000-10,000 cells would require from 100-500 million reads per sample). The T or B cell specific V(D)J assay requires 5000 reads per cell. Please contact us at epigenomicscore@med.cornell.edu to discuss the appropriate number of reads required for your experiments.
Data Processing
The sequencing data will be demultiplexed and post-processed using custom pipelines provided by 10x Genomics.
Single Cell Epigenomics
Assay description
We are currently offering Single Cell ATAC (NextGEM) as well as Epi Multiome ATAC + Gene Expression
Single cell assays require prior consultation and coordination with the core. Please contact us at epicore-sc-coord@med.cornell.edu at least three weeks before dropping off your samples to schedule a time.
The Chromium Single Cell ATAC (Assay for Transposase Accessible Chromatin) Solution accelerates the understanding of the regulatory landscape of the genome, thereby providing insights into cell variability. The high-throughput chromatin profiling of single cells in parallel allows researchers to see how chromatin compaction and DNA-binding proteins regulate gene expression at high resolution.
Sample Requirement:
- The total number of nuclei required in the suspension used as input is determined by the desired cell recovery target (between 500-10,000 cells); required sequencing read depth depends on the desired cell target. Chromium recommends 25,000 reads per nuclei (Example: 4 samples at 10,000 cells can be sequenced on one NovaSeq6000 SP flowcell, sequencing recipe 50-8-16-50)
- Given the variety of cells and sample types, general guidelines for sample preparation need to be optimized by each customer. Please check the relevant Sample Prep Guides for information on how to prepare cells.
- We can process up to 4 samples at a time and can only accept samples from local/internal clients.
| Service | Internal Price |
|---|---|
| Single Cell Multiome ATACseq & Gene Expression Library Prep | $4000 |
| Single Cell ATAC Library Prep | $2412 |
| Next GEM Chip J | $320 |
| Next GEM Chip H | $325 |
| NovaSeq X (100 cycles, 1.25B reads) + Bioinformatics Processing | $996 |
(Please Note: For Single Cell Multiome you require two different sequencing recipes, one for single cell RNAseq, one for single cell ATACseq)
Data Processing
The sequencing data from Chromium Single Cell ATAC will be demultiplexed and post-processed using custom pipelines provided by 10x Genomics.
The sequencing data from our Single Cell RRBS assays will be demultiplexed and post-processed in the same manner as our RRBS samples. Please click on the ERRBS section in the right hand menu for details.
Chromium Long Range Sequencing
Assay description
We are currently offering 10x Genomics applications for our long range genomic assay.
The Chromium Long Range Genome Solution provides long range information on a genome-wide scale, including variant calling, phasing and extensive characterization of genomic structure. Applications include interrogating heterogeneous cell populations, resolving phasing information and detecting structural variations. Please contact us at epigenomicscore@med.cornell.edu for further information and to discuss the details of your project.
Data Processing
The sequencing data will be demultiplexed and post-processed using custom pipelines provided by 10x Genomics.
Visium Spatial Transcriptomic Profiling
Assay Description
We are currently offering Visium CytAssist Spatial Transcriptomics, H&E workflow for mouse and human tissue samples. This assay allows mapping of the transcriptome within morphological context.
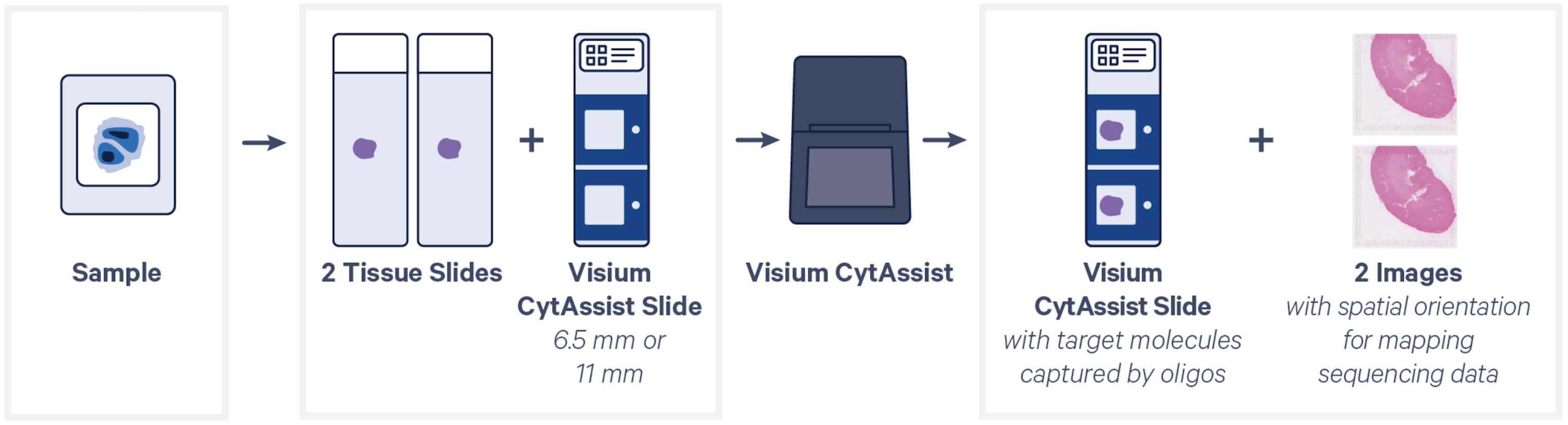
Spatial Transcriptomics assays require prior consultation and coordination with the core.
Please contact us at epigenomicscore@med.cornell.edu to discuss your project.
Sample Requirements
- RNA quality control (recommended DV200 > 30% for FFPE, DV200 > 50% for FxF, RIN > 4 for FF, RIN > 7 for FF HD 3’)
- A preliminary H&E staining should be performed to determine the region of interest.
- Samples can be FFPE, FF, or FxF tissue sections mounted on recommended histology slides (Tissue Slides). For a standardized workflow at the Epigenomics Core, we use Fisherbrand SuperFrost Plus slides (Cat# 12-550-15), while Schott Nexterion Slide H - 3D Hydrogel Coated (PN-1800434) is preferred for TMAs or tissues prone to detachment. Other validated options are listed in the 10x Genomics planner files CG000698 (HD), and CG000803 (HD 3’). Note that the Visium HD 3’ workflow is restricted to FF samples.
- The region of interest (6.5x6.5mm or 11x11mm) must be within an allowable area on the Tissue Slide.
- For sections thickness, sectioning protocol, tissue placement, size and location of the allowable area on the slide, tissue slides drying, storing and transfer to the Epigenomics Core, please refer to the Visium CytAssist Spatial Transcriptomics [PDF] infographic guide.
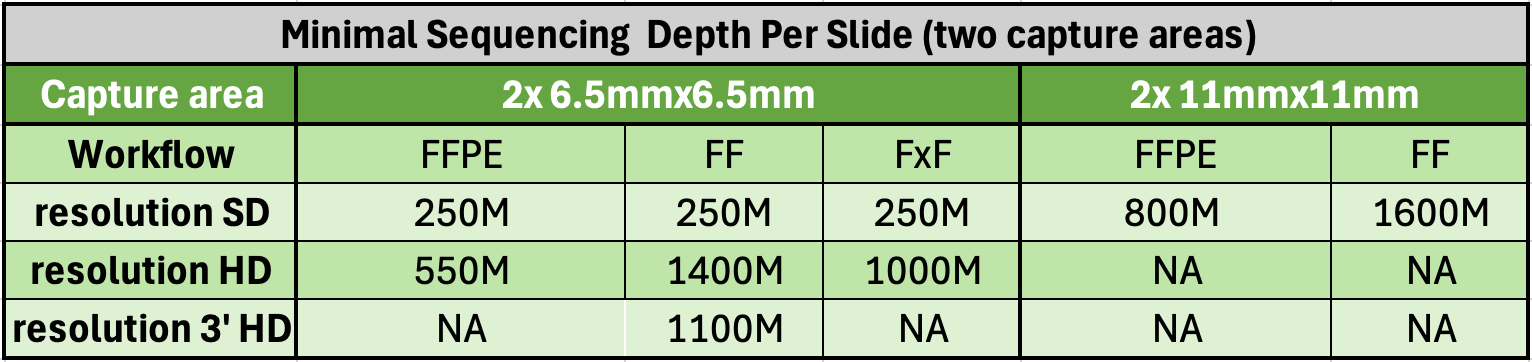
| Library Preperation + Sequencing for 1 Visium slide (2 capture areas per slide) |
Internal Price (WCM, WCM Qatar & Cornell U) |
|---|---|
| Visium CytAssist HD 6.5x6.5mm | $9237 |
| Visium CytAssist HD 11x11mm | $13375 |
| Visium CytAssist 3' HD 6.5x6.5mm | $9996 |
Submit a minimum of 2 tissue slides, or multiples of 2. Multiple tissues may be placed on a tissue slide, provided they fit within the 6.5x6.5mm (or 11x11mm) “Region of Interest” area.
Data Processing
The sequencing data from Visium Spatial Gene Expression will be demultiplexed and post-processed along with microscope generated images using the spaceranger software provided by 10x Genomics.
CosMx Single Cell Spatial Profiling
Assay Description
We are currently offering multiomic single-cell spatial profiling via the CosMx Spatial Molecule Imager from NanoString for FFPE and Fresh Frozen samples/slides. This assay allows high plex profiling of transcriptomics and proteomics within morphological context at sub-cellular resolution.
Single cell spatial profiling assays require prior consultation and coordination with the core. Please contact us at epigenomicscore@med.cornell.edu at least three weeks before dropping off your samples to schedule a time.
Sample Requirements
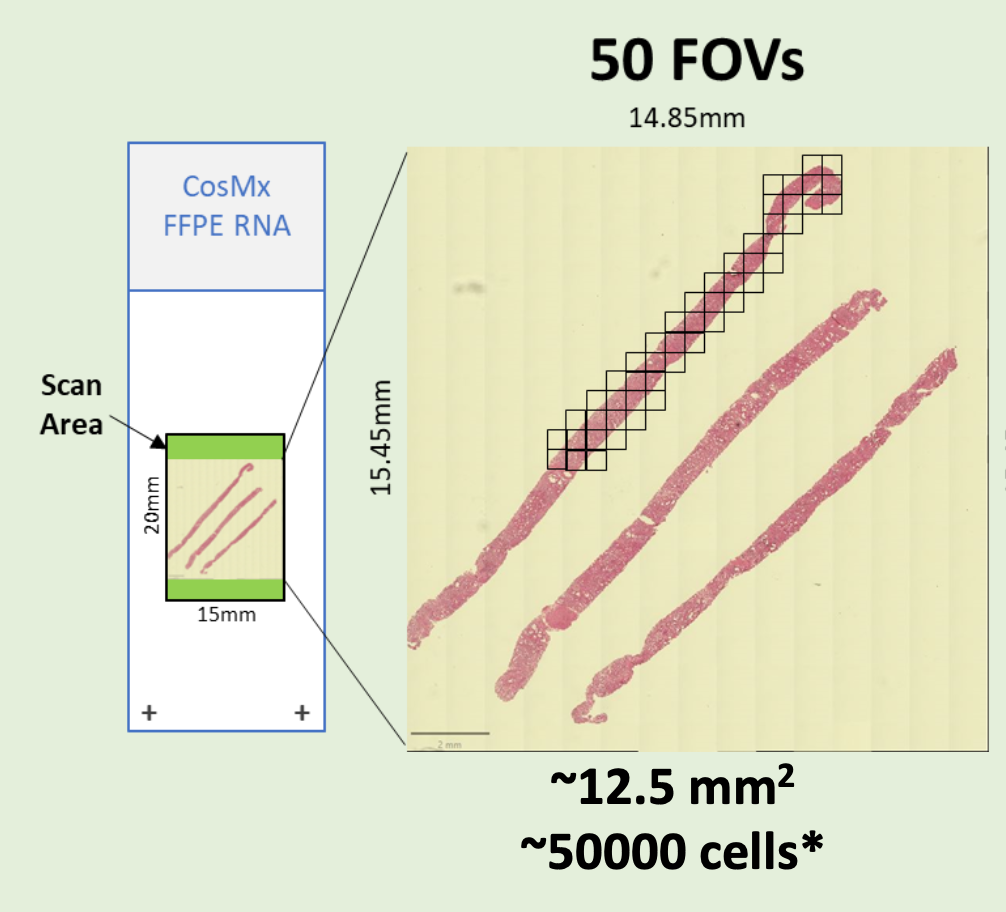
- FFPE microtome (RNA and Protein) or fresh frozen (RNA) sections on Nanostring-validated positively charged histology slides are required. Please review this detailed PDF to plan your CosMx experiment
- Preliminary sectioning, and H&E staining on parallel sections are required to determine the regions of interest and assign the prospective “Field of View” (FOVs) to be scored. Each “Field of View” (FOV) corresponds to a single CosMx 23x/1.1 NA objective acquisition area. FOVs are square-shaped, with the sides parallel to the slide edges. (It is not possible to rotate the FOVs to better fit the regions of interest.)
- Please submit a minimum of 2 histology slides, or multiples of 2.
- CosMx Planning Checklist [PDF]
| CosMx Panel Type | Internal Price (WCM, WCM Qatar & Cornell U) Per Slide | |
|---|---|---|
| Short Run (< 5 days)* | Long Run (> 5 days)* | |
| CosMx 1000x panel |
$6,054 (Up to 700 FOVs per run) |
$6,654 (700 to 1,500 FOVs per run) |
| CosMx 6000x (human) panel |
$8,637 (Up to 300 FOVs per run) |
$9,237 (300 to 1,200 FOVs per run) |
| CosMx WTX (human) panel | Not available |
$19,000 (Up to 800 FOVs per run) |
| CosMx-Protein 64x plex |
$4,820 (Up to 1,500 FOVs per run) |
Not available |
* Total run time depends directly on the specific panel used and the total number of FOVs selected per run.
Data Processing
CosMx spatial imaging data will be post-processed via the AtoMx Spatial Informatics Platform (SIP) provided by Bruker (Nanostring). Cell segmentation and Foundational pipelines that normalize, dimensionally reduce, and perform recommended downstream analysis will be run in AtoMx and you will get access to view, explore, and/or export your data via this SIP. Please refer to the CosMx Data Analysis Manual for further details about the post-processing and export of your CosMx/AtoMx data.
We will also share the following files (convenient for consumption by third-party tools) via our data portal PubShare:
Seurat Object - The .rds file is a Serurat Object (that includes polygon coordinates, metadata and dimensional reduction outputs).
Flat Files - The .tgz file is a tar-gzipped archive of flat files from the AtoMx pipeline export. Included flat files are as follows:
Count matrix file (cells with gene counts)
Cell metadata file (local (within the FOV)/global (within the flow cell) cell coordinates, morphology marker intensities)
Global transcripts file (global coordinates for every individual transcript on the flow cell; not applicable for protein studies)
Global cell boundaries file (global coordinates for the vertices of the polygon that represents the cells' boundaries - a representation of the cell segmentation)
Global FOV position file (coordinates of each FOV center)
Please note that your PubShare account is separate from your Agilent iLab account (used to create your service request) as well as your account to access the AtoMx SIP. If you don't already have access to the AtoMx SIP, expect an email from Okta (noreply@okta.com) to set up your account for AtoMx. Please be advised that your AtoMx account set-up link from Okta expires a week after it is sent.
Xenium Single Cell Spatial Profiling
Assay Description
The Epigenomics core offers in situ spatial gene expression profiling using the Xenium Analyzer (10x Genomics) for FFPE and fresh frozen (FF) human and mouse tissue sections. Xenium detects RNA (and optionally proteins) directly within intact tissue at subcellular resolution, preserving the spatial context of gene expression across the full tissue section. Two chemistries are available: Xenium v1 (pre-designed and custom panels up to 480 genes) and Xenium Prime (pre-designed 5K panels and standalone custom panels up to 2,000 genes).
- Subcellular resolution: Padlock probe-based in situ detection of RNA targets directly in FFPE or FF tissue sections.
- Spatial multi-omics: Optional cell segmentation available for both chemistries. Optional simultaneous protein detection of up to 23 immune, checkpoint, and tumor markers (v1 chemistry, Human FFPE only — Cell Segmentation required).
- Flexible panel design: Pre-designed (human and mouse) and custom panels available. Custom panels are ordered through the 10x Genomics Xenium Panel Designer; please account for additional lead time (6-8 weeks).
- Tissue microarray (TMA) compatible
Single cell spatial profiling assays require prior consultation and coordination with the core. Please contact us at epigenomicscore@med.cornell.edu at least three weeks before dropping off your samples to schedule a time. Requests are submitted and tracked through Agilent CrossLab/iLab.
Sample Requirements
FFPE or FF tissue sections must be placed on Xenium slides (10x Genomics) by personnel experienced in histology. The Epigenomics core does not provide this service. Slides can be ordered directly by the user, through a histology core facility, or via the Epigenomics core. Section thickness must be 5 µm (FFPE) or 10 µm (FF). The tissue must fit within the Xenium Sample Area (10.45 mm × 22.45 mm) without covering the fiducial markers. Slides must be submitted in pairs (or multiples of two). A DV200 ≥ 30% is recommended for FFPE samples. A preliminary H&E on a serial section on an uncharged slide is strongly recommended.
Please review this detailed PDF to plan your Xenium experiment
| Xenium Panel Type | Internal Price Per Slide (WCM, WCM Qatar & Cornell U) |
|---|---|
| Xenium V1 | |
| Xenium V1 - Up to 322-plex | $5,665 |
| Xenium V1 - Up to 380-plex | $5,842 |
| V1 Cell Segmentation | $945 |
| First Protein Subpanel | $814 |
| Any extra Protein Subpanel (each) | $210 |
| Xenium Prime (5K-plex) | |
| Xenium Prime | $9,580 |
| Prime Cell Segmentation | $667 |
For Custom panels prices please contact the 10x Genomics local sales representative.
Submit a minimum of 2 histology slides, or multiples of 2.
Data Processing
Xenium processes data in parallel with imaging and biochemistry cycles via Xenium Onboard Analysis. The Xenium data output files will be made available via PubShare for downstream exploration and analysis.
RNASeq (transcriptome analysis)
To continue availing of this service, please contact Dr. Jenny Xiang at the Genomics Resources Core Facility (GRCF).
[GRCF Service Request Portal]
To understand the dynamic state of the cell transcriptome, the core provides a range of RNA sequencing library preparations that can assess total RNA, mRNA and noncoding RNA in higher eukaryotes:
-
TruSeq Stranded mRNA:
polyA selection followed by library preparation is the standard and most widely used method for quantifying mRNA expression. Our protocol retains the strand orientation of the transcripts. [MORE]
Sample Requirement: Submit a minimum of 250 ng of purified RNA, at a concentration of 50 ng/ul; A260/280 > 2, picogreen quantification or bioanalyzer trace, RNA integrity number (RIN) >8.0
Epigenomics Core Quality Control: RNA Nano Bioanalyzer chip to determine RIN number and picogreen quantification.
-
Ultra Low Input RNA:
mRNA profiling of very low amounts of input material (<20ng) that is based on polyA selection using the Clontech SMArter technology. Often used for clinical samples or expression profiling of few cells (e.g. neurons, stem cells).
Sample Requirement: Submit a minimum of 20 ng of purified RNA, at a concentration of 5 ng/ul; A260/280 > 2, picogreen quantification or bioanalyzer trace, RNA integrity number (RIN) >8.0 OR 1000 cells in 10ul of Clontech 1X Single Cell Lysis buffer (cat # 635013)
Epigenomics Core Quality Control: RNA Pico Bioanalyzer chip to determine RIN number and picogreen quantification.
-
TruSeq Stranded Total RNA:
rRNA depletion followed by strand-specific library preparation. Generally more applicable towards simultaneous profiling of non-coding transcripts and mRNA although requires more input material. [MORE]
Sample Requirement: Submit a minimum of 250 ng of purified RNA, at a concentration of 50 ng/ul; A260/280 > 2, picogreen quantification or bioanalyzer trace, RNAs with an RNA integrity number (RIN) < 8 can be used
Epigenomics Core Quality Control: RNA Nano Bioanalyzer chip and picogreen quantification.
-
TruSeq RNA [DISCONTINUED]:
Please use TruSeq-stranded mRNA instead.
Agilent's Guide to Interpreting Bioanalyzer Results
Some studies that have used our RNASeq service: Kuo PY. et al. 2014, Satyaki PR. et al. 2014, Pimentel H. et al. 2014
| Service | Internal Price (WCM, WCM Qatar & Cornell U) |
External Academic Price |
|---|---|---|
| TruSeq RNA-seq (polyA) Library Prep | 185 | 300 |
| TruSeq Stranded RNA-seq (polyA) Library Prep | 170 | 225 |
| Ultra Low Input RNA-seq (polyA) Library Prep | 280 | 370 |
| TruSeq Total RNA-seq (stranded) Library Prep | 300 | 400 |
| HiSeq SR50 Single Read Clustering and 1 x 50 Sequencing Cycles | 1100 | 1600 |
| HiSeq PE50 Paired End Clustering and 2 x 50 Sequencing Cycles | 1650 | 2200 |
Sequencing depth required depends on your application.
For human transcriptome differential gene expression we recommend 4 samples per lane of SR50 sequencing.
For rare splicing events, translocations and some other experiments PE50 sequencing is recommended.
To calculate depth required for your custom application, please use the Illumina coverage calculator.
Sequence data (base call files or bcl files) generated from the sequencer are demultiplexed and converted to FASTQ files using the Illumina bcl2fastq software.
Your raw data will be available for download as a tar compressed archive (Sample_*.tar) of gzipped FASTQ files for each sample. Raw data can be post-processed upon request.
FASTQ files generated as described above are adapter trimmed and aligned to genomes available via Illumina's iGenome using the STAR aligner. Only raw reads that pass Illumina's purity filter are aligned. This pipeline results in the following file types:
*.bam - Upon alignment (if requested) the aligned data processed by STAR aligner is in the widely accepted BAM format (a binary version of the SAM format).
*.bai - This is an index file for your BAM alignments and allows certain browsers (such as the IGV browser) to better view the .bam file.
*-SJ.out.tab - The high confidence collapsed splice junctions in tab-delimited format. Only junctions supported by uniquely mapping reads are reported.
*-Log.final.out - A text file containing the STAR aligner generated summary statistics for the alignment of each sample.
Bioinformatics Support
The Epigenomics Core Facility provides data analysis services and consultation on a per project basis. These include but are not limited to:
- RNASeq differential expression
- RRBS differential methylation
- ChIPSeq peak calling and differential binding
- Functional annotation and analysis
- Single Cell and Spatial Profiling
- Custom alignments
We require that publications and/or presentations enabled by custom data analysis through core personnel contain proper author credit for the bioinformatician. Please discuss this with us before publication/presentation.
Please note that some bioinformatics support is included in the price for some of our services. For example:
- Quality control statistics for your samples
- 2 years data storage on our backed up servers with unlimited password protected access for sequencing projects
- Illumina iGenome alignments (if available and when requested for internal customers)
- Methylation calling for DNA methylation sequencing projects (where genomes are available)
- Methylation ratios for Epityper mass array projects
- Splice junction tables for RNASeq alignments
Please review the Data Analysis and Retrieval section of our Frequently Asked Questions page for further details.
| Service Type | Internal Price (WCM, WCM Qatar & Cornell U) | External Academic Price |
|---|---|---|
| Data analysis support / hour | $125 | $175 |
Related Publications:
- Garman, B. et al. Front Immunol. 2023 Sep 19:14:1151748. [PubMed]
- Singh, N. et al. Nat Commun. 2021 Dec 21;12(1):7349. [PubMed]
- Singh, P. et al. Sci Rep. 2021 Oct 26;11(1):21107 [PubMed]
- Singh, P. et al. Ann N Y Acad Sci. 2020. 1490(1):42-56 [PubMed]
- Zhang, T. et al. Blood Cancer J. 2017. Sep 8; 7(9), e606 [PubMed]
- Bayliss, J., Mukherjee P. et al. Sci Transl Mel 2016. Nov 23; 8 (366), 366ra161 [PubMed]
- Garrett-Bakelman, FE. Sheridan, C. et al. J Vis Exp. 2015. Feb 24; (96) [PubMed]